Наследственные болезни
I
Насле́дственные боле́зни
заболевания человека, обусловленные хромосомными и генными мутациями. Нередко ошибочно термины «наследственная » и «врожденная болезнь» употребляются как синонимы, однако врожденными болезнями называют те заболевания, которые имеются уже при рождении ребенка и могут быть обусловлены как наследственными, так и экзогенными факторами. Таковы, например, связанные с воздействием на и плод ионизирующего излучения, химических соединений, лекарственных средств, принимаемых матерью, а также внутриутробных инфекций. Однако далеко не все Н.б. относят к врожденным, поскольку многие из них проявляются уже после периода новорожденности (например, Гентингтона клинически обнаруживается после 40 лет). Наследственные и врожденные болезни являются причиной госпитализации детей почти в 30% случаев и даже больше (с учетом болезней неизвестной природы, которые в значительной степени могут быть вызваны генетическими факторами). В качестве синонима термина «наследственные болезни» не следует также рассматривать термин «семейные болезни», т.к. семейные заболевания могут быть обусловлены не только наследственными факторами, но и условиями жизни, национальными либо профессиональными традициями семьи. К болезням второй группы относят так называемые мультифакториальные болезни, в основе которых лежит взаимодействие генетических и средовых факторов. К болезням этой группы относятся , атеросклероз, желудка и двенадцатиперстной кишки, сахарный , аллергические заболевания, многие пороки развития, определенные формы ожирения. Генетические факторы, представленные определенной полигенной системой, обусловливают генетическую предрасположенность, которая может быть реализована при воздействии неблагоприятных или вредных факторов окружающей среды (физического или умственного переутомления, нарушения режима и сбалансированности питания и т.п.). Для одних из них влияние окружающей среды имеет большее, для других - меньшее значение. К мультифакториальным болезням относят также состояния, при которых роль генетического фактора может играть один единственный мутантный , но проявляется это состояние также только при определенных условиях. Примером такого состояния может служить дефицит глюкозо-6-фосфат - . Болезни третьей группы связаны исключительно с воздействием неблагоприятных или вредных факторов окружающей среды, в их возникновении практически не играет никакой роли. К этой группе относят , ожоги, острые . Однако генетические факторы могут оказывать определенное влияние на течение патологического процесса, т. е. на темпы выздоровления, переход острых процессов в хронические, развитие декомпенсации функций пораженных органов. Наследственные болезни обычно подразделяют на три основные группы: моногенные, полигенные (мультифакториальные, или болезни с наследственным предрасположением) и хромосомные. Клиническая классификация Н.б. построена по органному и системному принципам. Согласно этой классификации, выделяют Н.б, нервной, эндокринной, дыхательной и сердечно-сосудистой систем, печени, желудочно-кишечного тракта, почек, системы крови, кожи, уха, носа, и др. Такая классификация в значительной степени условна, т.к. большинство Н.б. характеризуется вовлечением в нескольких органов или системным поражением тканей. Моногенные болезни
по типу наследования могут быть аутосомно-доминантными, аутосомно-рецессивными и сцепленными с полом; по фенотипическому проявлению - ферментопатиями (болезнями обмена веществ, в т.ч. болезнями, обусловленными нарушением репарации ): болезнями, обусловленными молекулярной патологией структурных белков; иммунопатологией, в т.ч. болезнями, вызванными нарушениями в системе комплемента; нарушениями синтеза транспортных белков (в т.ч. белков крови) и пептидных гормонов; патологией свертывающей системы крови; дефектами механизма переноса веществ через клеточные мембраны. Среди моногенных болезней также выделяют группу синдромов с множественными врожденными пороками развития, при которых неуточнен первичный мутантного гена. Моногенные болезни наследуются в полном соответствии с законами Менделя (см. Наследственность).
Большинство известных Н.б. обусловлено мутациями структурных генов (см. ), возможность этиологической роли мутаций генов-регуляторов при некоторых заболеваниях до сих доказана лишь косвенно. При аутосомно-рецессивном типе наследования мутантный ген проявляется только в гомозиготном состоянии. Больные мальчики и девочки рождаются с одинаковой частотой. Вероятность рождения больного ребенка составляет 25%. Родители больных детей фенотипически могут быть здоровы, но являются гетерозиготными носителями мутантного гена. Аутосомно-рецессивный тип наследования более характерен для заболеваний, при которых нарушена одного или нескольких ферментов, - так называемый ферментопатий (Ферментопатии).
Ферментопатии, относящиеся к моногенным Н.б. по фенотипическому проявлению, составляют наиболее обширную и лучше всего изученную группу Н.б. Первичный дефект фермента расшифрован более чем для 200 . Возможны следующие причины ферментопатии: а) не синтезируется совсем; б) в молекуле фермента нарушена последовательность аминокислот, т.е. изменена его первичная структура; в) отсутствует или неправильно синтезируется соответствующего фермента; г) фермента изменена в связи с аномалиями в других ферментных системах; д) фермента обусловлена генетически детерминированным синтезом веществ, вызывающих инактивацию этого фермента. Мутация гена может повлечь за собой нарушение синтеза белков, выполняющих пластические (структурные) функции. Нарушение синтеза структурных белков - вероятная причина таких заболеваний, как Остеодисплазии и Остеогенез несовершенный.
Есть данные об определенной роли этих нарушений в патогенезе наследственных нефритоподобных заболеваний - синдрома Альпорта и семейной гематурии. ткани в результате аномалий в структуре белков может наблюдаться не только в почках, но и в любых других органах. структурных белков характерна для большинства Н.б., наследуемых по аутосомно-доминантному типу. Мутация гена может привести к развитию болезней, вызванных иммунодефицитными состояниями (см. Иммунопатология).
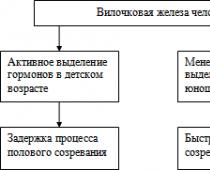
Наиболее тяжело протекает , особенно в сочетании с аплазией вилочковой железы. Причиной появления гемоглобина с аномальной структурой при серповидно-клеточной является замена в его молекуле остатка глутаминовой кислоты на остаток валина. Подобная заменю является результатом генной мутации. Это открытие послужило началом интенсивного изучения большой группы Н.б., названных гемоглобинопатиями (Гемоглобинопатии).
Известен ряд мутантных генов, контролирующих факторов свертывания крови (см. Свертывающая крови (Свёртывающая система крови)).
Генетически детерминированные нарушения синтеза антигемофилического глобулина (фактор VIII) приводят к развитию гемофилии А. При нарушении синтеза тромбопластического компонента (фактор IX) развивается гемофилия В. Недостаток предшественника тромбопластина лежит в основе патогенеза гемофилии С. Генные мутации могут быть причиной нарушения механизма транспорта различных соединений через клеточные мембраны. Наиболее изучены наследственная транспорта аминокислот (Аминокислоты) в кишечнике и почках, глюкозы и галактозы (см. Углеводы),
отмечены генетически обусловленные нарушения нормального функционирования так называемый К + . Na + -иoннoro насоса (АТФ-азы) клетки. Известны заболевания, причиной которых служит дефект механизмов, ответственных за поддержание нормального градиента концентраций ионов К + и Mg 2+ по обе стороны клеточной мембраны, что клинически проявляется периодическими приступами тетании (Тетания).
Примером заболевания, вызванного генетически обусловленным дефектом механизма транспорта аминокислот через клеточные мембраны, является , клиническим выражением которой является и симптомы пиелонефрита. Классическая цистинурия обусловлена нарушением переноса ряда диаминокарбоновых кислот (аргинина, лизина) и цистина через клеточные мембраны как в кишечнике, так и в почках. Патология реабсорбции глюкозы в почечных канальцах - почечная - связана с нарушением функции мембранных белков-переносчиков или с дефектами в системе обеспечения энергией процессов активного транспорта глюкозы; наследуется по аутосомно-доминантному типу. Нарушение реабсорбции бикарбонатов в проксимальных отделах почечных канальцев или нарушение секреции водородных ионов клетками почечного эпителия дистальных отделов почечных канальцев лежит в основе развития двух типов почечного канальцевого ацидоза (см. Кислотно-щелочное равновесие).
Частота встречаемости моногенных Н.б. колеблется у разных этнических групп вразных географических зонах. В странах Западной Европы и СССР наиболее распространенными Н.б. являются (1:20 000 - 1:40 000), (1: 17 000), (1:12 000 - 1:15 000), цистинурия (1:14 000), (1:1200 - 1:5000). Частота встречаемости гиперлипопротеинемий (в т.ч. и полигенно наследуемых форм) достигает 1:100 - 1:200. К часто встречающимся Н.б. относятся гемофилия (1:10 000; болеют мальчики), (1:7000), мальабсорбции (1:3000), (1:5000 - 1:11 000). Частота встречаемости значительного числа Н.б. обмена пока не установлена, хотя это не свидетельствует об их редкости. Полигенные (мультифакториальные) наследственные болезни, или болезни с наследственным предрасположением
, обусловлены взаимодействием нескольких (или многих) генов в полигенных системах и факторов окружающей среды. болезней с наследственным предрасположением, несмотря на их распространенность, изучен недостаточно. Большое значение имеет поиск фенотипических маркеров наследственной предрасположенности к определенному заболеванию, например, аллергический может быть диагностирован на основании повышенного содержания в крови иммуноглобулина Е и повышенной экскреции с мочой минорных метаболитов триптофана. Определены биохимические маркеры наследственной предрасположенности к сахарному диабету (повышенная к глюкозе, содержание в крови иммунореактивного инсулина), конституционально-экзогенному ожирению, гипертонической болезни (гиперлипопротеинемия). Достигнуты успехи в изучении взаимосвязи между группами крови АВО, антигенами системы гистосовместимости HLA и возможностью развития некоторых болезней. Установлено, что для лиц с тканевым гаплотипом HLA В8 высок риск заболевания хроническим гепатитом, целиакией и миастенией; для лиц с гаплотипом HLA А2 - хроническим гломерулонефритом, лейкозом; для лиц с гаплотипом HLA DW4 - ревматоидным артритом, для лиц с гаплотипом HLA А1 - атопической аллергией. с системой HLA обнаружена примерно для 90 заболеваний человека, многие из которых характеризуются нарушениями системы иммунитета. Болезни с наследственным предрасположением также имеют особенности распространения в разных странах. Так, по данным Шандса (A.R. Shands), частота расщепления и нёба в Англии составляет 1:515, в Японии - 1:333, а врожденный бедра в Японии наблюдается в 10 раз чаще, чем в Англии. Хромосомные болезни
подразделяют на аномалии, обусловленные изменениями количества хромосом (полиплоидии, анэуплоидии) или структурными перестройками хромосом - делециями, инверсиями, транслокациями, дупликациями. Хромосомные мутации, возникшие в зародышевых клетках (гаметах), проявляются в так называемых полных формах. (Хромосомы) и структурные изменения, появившиеся на ранних стадиях дробления зиготы, ведут к развитию мозаицизма. Вероятность появления хромосомных аберраций резко увеличивается в гаметах женщин старше 35 лет. Частота встречаемости всех хромосомных болезней среди новорожденных, по данным Кэбека (М.М. Kaback), составляет 5,6:1000, при этом все виды анэуплоидий, включая мозаичные формы, составляют 3,7:1000, трисомии по аутосомам и структурные перестройки - 1,9:1000. Половину всех случаев структурных перестроек хромосом представляют семейные случаи, все трисомии являются спорадическими случаями, т. е. следствием вновь возникших мутаций. По данным Полани (P. Polani), около 7% всех беременностей осложнены хромосомными аберрациями плода, которые в подавляющем большинстве случаев ведут к спонтанным абортам. Диагноз ряда Н.б. не представляет существенных затруднений и основывается на данных, полученных в результате общего клинического обследования (например, болезни Дауна, гемофилии, гаргоилизма, адреногенитального синдрома). Однако в большинстве случаев при диагностике Н.б. возникают серьезные затруднения в связи с тем, что многие из этих болезней по клиническим проявлениям очень сходны с приобретенными болезнями - так называемые фенокопиями Н.б. Известно также существование ряда фенотипически сходных, но гетерогенных в генетическом отношении болезней (например, синдром Марфана и , галактоземия и синдром Лоу, фосфат-диабет и почечный канальцевый ). Все случаи атипично протекающих или хронических заболеваний требуют клинико-генетического анализа. На наследственную болезнь может указывать наличие специфических клинических признаков. Среди них особое диагностическое значение могут иметь признаки дисплазии - эпикант, седловидный , особенности строения лица («птичье», «кукольное», олигомимичное и др.), черепа ( , брахицефалия, «ягодичная» форма черепа и др.), глаз, зубов, конечностей и др. При подозрении на Н.б. генетическое начинают с получения подробных клинико-генеалогических данных на основании результатов его опроса о состоянии здоровья ближайших и отдаленных родственников, а также специального обследования членов семьи, что позволяет составить медицинскую родословную больного и определить наследования патологии. Вспомогательное, а в ряде случаев решающее диагностическое значение имеют данные, полученные с помощью различных параклинических методов, в т.ч. биохимических, и цитогенетических и молекулярно-генетических исследований. Разработаны биохимические методы диагностики нарушений обмена веществ, основанные на применении хроматографии (Хроматография), электрофореза, ультрацентрифугирования и т.д. Для диагностики заболеваний, вызванных недостаточностью ферментов, применяют методы определения активности этих ферментов в плазме и клетках крови, в материале, полученном при биопсии органов. Биохимические исследования проводят также на клетках, выращенных в культуре, что позволяет обнаружить ряд болезней, обусловленных нарушением обмена липидов, гликозаминогликанов (мукополисахаридов), углеводов, аминокислот, нуклеиновых кислот, - всего более 100 заболеваний. Наиболее разработанными являются методы диагностики болезни Тея - Сакса (см. Амавротическая идиотия),
синдрома Леша - Найхана (см. Подагра),
некоторых мукополисахаридов (Мукополисахариды).
Проведение биохимических исследований при Н.б. обмена в ряде случаев требует применения нагрузочных проб соединениями, которых, как предполагают, нарушен. Однако сложные аналитические методы не могут быть использованы для массовых обследований. В связи с этим при массовых осмотрах проводят двухэтапное обследование с применением на начальном этапе простых полуколичественных методов и (при положительных результатах обследования на первом этапе) - более сложных аналитических методов, выполнение которых возможно лишь в специальных центрах. Эти программы получили название просеивающих, или скрининг-программ. Внедрение методов автоматического биохимического анализа облегчает проведение массового обследования детей на Н.б. Массовые обследования детских контингентов (особенно контингентов новорожденных) позволяют выявлять наследственные нарушения обмена в доклинической стадии, когда и соответствующее медикаментозное способны полностью предупредить развитие тяжелой инвалидности. Разработка новых методов культивирования клеток, биохимические и цитогенетическое исследования сделали возможной пренатальную диагностику многих Н.б., в т.ч. всех хромосомных болезней и болезней, сцепленных с Х-хромосомой, а также целый ряд генетически обусловленных нарушений обмена веществ. Результаты подобных исследований могут служить показанием для прерывания беременности или начала лечения аномалий обмена еще во внутриутробном периоде. Пренатальная Н.б. показана в тех случаях, когда у одного из родителей обнаруживается структурная перестройка хромосом ( , инверсия), когда беременных женщин превышает 35 лет и когда в семье прослеживаются доминантно наследуемые заболевания или существует высокий риск возникновения рецессивных наследственных болезней - аутосомных или сцепленных с Х-хромосомой. Для антенатальной диагностики используют также рентгенографию и ультразвуковое исследование плода. Лечение и наследственных болезней
. В большинстве случаев Н.б. проводится симптоматическое лечение, направленное на коррекцию отдельных проявлений. Одним из наиболее распространенных методов патогенетического лечения Н.б. обмена является диетотерапия. Проведение диетотерапии требует строгого соблюдения ряда условий: точного диагноза аномалии обмена, исключающего ошибки, связанные с существованием фенотипически сходных синдромов; максимальной адаптации диеты к потребностям растущего организма; тщательного клинического и биохимического контроля. Наиболее полно изучены возможности диетической коррекции обмена фенилаланина при фенилкетонурии. Для диетической коррекции галактоземии созданы специальные препараты. Препараты типа энпита с успехом применяют при лечении других Н.б. (синдрома Марфана, синдрома Лоренса - Муна - Бидля). Предложены также специальные диеты для лечения гистидинемии, гомоцистинурии, кетоацидурии и др. Продолжается поиск методов лечения больных с наследственными ферментопатиями. Заместительная , при ферментопатиях, в основе которых лежит недостаточность ферментов или их проферментов, участвующих в процессах расщепления и всасывания (муковисцидозе, недостаточности дисахаридаз, трипсиногена и др.), заключается в приеме желудочного сока, препаратов пепсина, трипсина, панкреатина; проходит клинические испытания использование лактазы, дрожжевой сахаразы, гамма-амилазы при нарушениях всасывания лактозы, сахарозы и крахмала, Заместительная терапия препаратами гамма-глобулина, обогащенными антителами или иммуноглобулинами других классов, проводится при лечении наследственных иммунопатологий, связанных с дефицитом иммуноглобулинов. Для лечения наследственных эндокринных заболеваний вводят кортйкостероиды (например, при адреногенитальном синдроме), препараты гормонов щитовидной железы (при гипотиреозе), (при сахарном диабете) и т.п. Основным препятствием при лечении наследственных ферментопатий методом введения недостающих ферментов, т.е. методом заместительной ферментотерапии, являются иммунные реакции на введение чужеродного белка. Новые возможности в этом направлении открывает использование искусственно созданных липидных частиц - липосом. Клетки тканей захватывают , под действием клеточных липаз оболочка липосомы разрушается и фермент получает возможность проявить свое действие внутри клетки. В качестве оболочки для вводимого с терапевтической целью фермента используют также больного, капсулы из нейлона. Новым направлением в лечении Н.б. является разработка методов индуцирования синтеза ферментов с помощью химических препаратов и гормонов. Так, установлено, например, что индуцируют синтез глюкуронилтрансферазы - фермента, необходимого для образования глюкуронидов Билирубин а (так называемого прямого билирубина), стероидных гормонов и ряда других соединений. Отмечено значительное повышение активности глюкуронилтрансферазы под влиянием фенобарбитала у больных с синдромом Криглера - Найяра, который характеризуется резкой гипербилирубинемией (Гипербилирубинемия) в связи с генетически обусловленной недостаточностью этого фермента. Глюкокортиконды активизируют синтез глюкозо-6-фосфат - дегидрогеназы и могут использоваться при лечении гликогеноза I типа (болезни Гирке) с целью предупреждения гипогликемических состояний и снижения интенсивности накопления гликогена в тканях (см. Гликогенозы).
Установлено индуцирующее влияние кортикостероидов на синтез и созревание ферментных систем кишечника, в частности дисахаридаз. Эстрогенные обусловливают увеличение концентрации церулоплазмина в крови, поэтому их используют при лечении гепатоцеребральной дистрофии. Индуцировать синтез ферментов могут и , причем это особенно заметно при так называемой витаминозависимых состояниях, которые характеризуются развитием гипо- или авитаминоза не в связи с ограниченным поступлением витаминов в , а в результате нарушения синтеза специфических транспортных белков или апоферментов (см. Ферменты).
Хорошо известна эффективность высоких доз витамина В 6 (от 100 мг
и выше в сутки) при так называемых пиридоксинзависимых состояниях и заболеваниях (цистатионинурии, гомоцистинурин, семейной гипохромной анемии, синдроме Клаппа - Комроуэра, болезни Хартнупа, некоторых формах бронхиальной астмы). Высокие дозы витамина D (до 50 000 - 200 000 в сутки) оказались эффективными при наследственных рахитоподобных заболеваниях (фосфат-диабете, синдроме Тони - Дебре - Фанкони, почечном канальцевом ацидозе). ВитаминС в дозах до 1000 мг
в сутки применяют при лечении алкаптонурии. Высокие дозы витамина А назначают больным с синдромами Гурлер и Гунтера (). Отмечено улучшение состояния больных мукополисахаридозами под влиянием преднизодона. Успехи пластической и восстановительной хирургии определили высокую эффективность хирургического лечения наследственных и врожденных пороков развития. В практику лечения Н.б. внедряются методы трансплантации, что позволит не только заменить органу, подвергшиеся необратимым изменениям, но и осуществлять пересадки с целью восстановления синтеза белков и ферментов, отсутствующих у больных. Большой интерес может представить иммунокомпетентных органов (вилочковой железы, костного мозга) при лечении разных форм наследственных иммунопатологий. Одним из методов лечения Н.б. является назначение препаратов, связывающих токсические продукты, образующиеся в результате блокирования определенных биохимических реакций. Так, для лечения гепатоцеребральной дистрофии (болезни Вильсона - Коновалова) применяют препараты, образующие растворимые комплексные соединения с медью (унитиол, пеницилламин). , специфически связывающие , находят применение при лечении гемохроматоза, а , образующие растворимые комплексные соединения кальция, - при лечении наследственных тубулопатий с нефролитиазом. При лечении гиперлипопротеинемий применяют холестирамин, который связывает в кишечнике и препятствует его реабсорбции. Успехи в профилактике и лечении Н.б. в первую очередь связаны с созданием диспансерного обслуживания больных с наследственными заболеваниями. В нашей стране организуются консультативные кабинеты по медицинской генетике и центры по
медико-генетическому консультированию (Медико-генетическое консультирование),
по наследственной патологии у детей и по пренатальной наследственной патологии.
От родителей ребенок может приобрести не только определенный цвет глаз, рост или форму лица, но и передающиеся по наследству. Какие они бывают? Как можно их обнаружить? Какая классификация существует?
Механизмы наследственности
Прежде, чем говорить о заболеваниях, стоит разобраться, что такое Вся информация о нас содержится в молекуле ДНК, которая состоит из невообразимо длинной цепочки аминокислот. Чередование этих аминокислот уникально.
Фрагменты цепочки ДНК называются генами. В каждом гене заключается целостная информация об одном или нескольких признаках организма, которая передается от родителей детям, например, цвет кожи, волос, черта характера и т. д. При их повреждении или нарушении их работы возникают генетические заболевания, передающиеся по наследству.
ДНК организовано в 46 хромосомах или 23 парах, одна из которых является половой. Хромосомы отвечают за активность генов, их копирование, а также восстановление при повреждениях. В результате оплодотворения в каждой паре присутствует одна хромосома от отца, а другая от матери.
При этом один из генов будет доминантным, а другой рецессивным или подавляемым. Упрощенно, если у отца ген, отвечающий за цвет глаз, окажется доминантным, то ребенок унаследует этот признак именно от него, а не от матери.
Генетические заболевания
Передающиеся по наследству болезни возникают, когда в механизме хранения и передачи генетической информации происходят нарушения или же мутации. Организм, чей ген поврежден, будет передавать его своим потомкам точно так же, как и здоровый материал.

В том случае, когда патологический ген является рецессивным, он может и не проявляться у следующих поколений, но они будут его переносчиками. Шанс, что не проявится, существует, когда здоровый ген тоже окажется доминантным.
В настоящее время известно больше 6 тысяч наследственных заболеваний. Многие из них проявляются после 35 лет, а некоторые могут никогда не заявить о себе хозяину. С крайне высокой частотой проявляется сахарный диабет, ожирение, псориаз, болезнь Альцгеймера, шизофрения и другие расстройства.
Классификация
Генетические заболевания, передающиеся по наследству, имеют огромное количество разновидностей. Для разделения их на отдельные группы может учитываться локация нарушения, причины, клиническая картина, характер наследственности.
Болезни могут классифицироваться по типу наследования и локации дефектного гена. Так, важно, расположен ген в половой или неполовой хромосоме (аутосоме), а также является он подавляющим или нет. Выделяют заболевания:
- Аутосомно-доминантные - брахидактилия, арахнодактилия, эктопия хрусталика.
- Аутосомно-рецессивные - альбинизм, мышечная дистония, дистрофия.
- Ограниченные полом (наблюдаются только у женщин или мужчин) - гемофилия А и Б, цветовая слепота, паралич, фосфат-диабет.
Количественно-качественная классификация наследственных болезней выделяет генные, хромосомные и митохондриальные виды. Последний относится к нарушениям ДНК в митохондриях за пределами ядра. Первые два происходят в ДНК, которая находится в ядре клетки, и имеют несколько подвидов:
Моногенные | Мутации или отсутствие гена в ядерной ДНК. | Синдром Марфана, адреногенитальный синдром у новорожденных, нейрофиброматоз, гемофилия А, миопатия Дюшенна. |
Полигенные | Предрасположенность и действие | Псориаз, шизофрения, ишемическая болезнь, цирроз, бронхиальная астма, сахарный диабет. |
Хромосомные |
||
Изменение структуры хромосом. | Синдромы Миллера-Диккера, Вильямса, Лангера-Гидиона. |
|
Изменение числа хромосом. | Синдромы Дауна, Патау, Эдвардса, Клайфентера. |
|
Причины возникновения
Наши гены склонны не только накапливать информацию, но и изменять её, приобретая новые качества. Это и есть мутация. Происходит она довольно редко, примерно 1 раз на миллион случаев, и передается потомкам, если произошла в половых клетках. Для отдельных генов частота мутации составляет 1:108.
Мутации являются естественным процессом и составляют основу эволюционной изменчивости всех живых существ. Они могут быть полезными и вредными. Одни помогают нам лучше приспособиться к окружающей среде и способу жизни (например, противопоставленный большой палец руки), другие приводят к заболеваниям.

Возникновение патологий в генах учащают физические, химические и биологические Таким свойством обладают некоторые алкалоиды, нитраты, нитриты, некоторые пищевые добавки, пестициды, растворители и нефтяные продукты.
Среди физических факторов находятся ионизирующие и радиоактивные излучения, ультрафиолетовые лучи, чрезмерно высокие и низкие температуры. В качестве биологических причин выступают вирусы краснухи, кори, антигены и т. д.
Генетическая предрасположенность
Родители влияют на нас не только воспитанием. Известно, что одни люди имеют больше шансов появления некоторых заболеваний, чем другие из-за наследственности. Генетическая предрасположенность к заболеваниям возникает, когда кто-то из родственников имеет нарушения в генах.
Риск возникновения конкретного заболевания у ребенка зависит от его пола, ведь некоторые болезни передаются только по одной линии. Он также зависит от расы человека и от степени родства с больным.
Если у человека с мутацией рождается ребенок, то шанс унаследования болезни будет 50%. Ген вполне может никак себя не проявить, будучи рецессивным, а в случае брака со здоровым человеком, его шансы передаться потомкам составят уже 25%. Однако если супруг тоже будет владеть таким рецессивным геном, шансы проявления его у потомков снова увеличатся до 50 %.
Как выявить болезнь?
Вовремя обнаружить заболевание или предрасположенность к нему поможет генетический центр. Обычно такой есть во всех крупных городах. Перед сдачей анализов проводится консультация с врачом, чтобы выяснить, какие проблемы со здоровьем наблюдаются у родственников.
Медико-генетическое обследование проводится путем взятия крови на анализ. Образец внимательно изучается в лаборатории на предмет каких-либо отклонений. Будущие родители обычно посещают подобные консультации уже после наступления беременности. Однако в генетический центр стоит прийти и во время её планирования.

Наследственные заболевания серьезно отражаются на психическом и физическом здоровье ребенка, влияют на продолжительность жизни. Большинство из них тяжело поддается лечению, а их проявление только корректируется медицинскими средствами. Поэтому лучше подготовиться к подобному ещё до зачатия малыша.
Синдром Дауна
Одна из наиболее распространенных генетических болезней - синдром Дауна. Она встречается в 13 случаях из 10000. Это аномалия, при которой человек имеет не 46, а 47 хромосом. Диагностировать синдром можно сразу при рождении.
Среди главных симптомов уплощенное лицо, приподнятые уголки глаз, короткая шея и недостаток мышечного тонуса. Ушные раковины, как правило, маленькие, разрез глаз косой, неправильная форма черепа.

У больных детей наблюдаются сопутствующие расстройства и болезни - пневмония, ОРВИ и т. д. Возможно возникновение обострений, например, потеря слуха, зрения, гипотериоз, заболевания сердца. При даунизме замедлено и часто остается на уровне семи лет.
Постоянная работа, специальные упражнения и препараты значительно улучшают ситуации. Известно много случаев, когда люди с подобным синдромом вполне могли вести самостоятельную жизнь, находили работу и достигали профессиональных успехов.
Гемофилия
Редкое наследственное заболевание, поражающее мужчин. Встречается один раз на 10 000 случаев. Гемофилия не лечится и возникает в результате изменения одного гена в половой Х-хромосоме. Женщины являются только переносчиками болезни.

Основной характеристикой является отсутствие белка, который отвечает за свертывание крови. В таком случае, даже незначительная травма вызывает кровотечение, которое не просто остановить. Иногда оно проявляет себя только на следующий день после ушиба.
Английская королева Виктория была носителем гемофилии. Она передала болезнь многим своим потомкам, в том числе и цесаревичу Алексею - сыну царя Николая II. Благодаря ей болезнь стали называть «царской» или «викторианской».
Синдром Ангельмана
Болезнь часто называют «синдромом счастливой куклы» или «синдромом Петрушки», так как у больных наблюдаются частые вспышки смеха и улыбки, хаотические движения рук. При данной аномалии характерно нарушение сна и психического развития.

Синдром возникает раз на 10 000 случаев из-за отсутствия некоторых генов в длинном плече 15-й хромосомы. Болезнь Ангельмана развивается только, если гены отсутствуют в хромосоме, доставшейся от матери. Когда те же гены отсутствуют в отцовской хромосоме, возникает синдром Прадера-Вилли.
Заболевание нельзя излечить полностью, но облегчить проявление симптомов возможно. Для этого проводятся физические процедуры и массажи. Полностью самостоятельными больные не становятся, но при лечении могут сами себя обслуживать.
У каждого здорового человека есть 6-8 поврежденных генов, однако они не нарушают функций клеток и не приводят к заболеванию, поскольку являются рецессивными (непроявляющимися). Если же человек наследует от матери и отца два сходных аномальных гена, он заболевает. Вероятность подобного совпадения чрезвычайно мала, но она резко возрастает, если родители являются родственниками (то есть имеют похожий генотип). По этой причине частота генетических аномалий высока в замкнутых группах населения.
Каждый ген в человеческом организме отвечает за выработку определенного белка. Из-за проявления поврежденного гена начинается синтез аномального белка, что приводит к нарушениям функций клеток и пороку развития.
Установить риск возможной генетической аномалии может врач, расспросив вас о заболеваниях родственников «до третьего колена» как с вашей стороны, так и со стороны вашего мужа.
Генетических заболеваний великое множество, причем некоторые - очень редкие.
Список редких наследственных болезней
Приведем характеристики некоторых генетических заболеваний.
Синдром Дауна (или трисомия 21) - хромосомное заболевание, характеризующееся умственной отсталостью и нарушением физического развития. Возникает заболевание вследствие наличия третьей хромосомы в 21-й паре (всего человек имеет 23 пары хромосом). Это самое распространенное генетическое заболевание, встречающееся примерно у одного из 700 новорожденных. Частота синдрома Дауна возрастает у детей, рожденных женщинами старше 35 лет. Больные этой болезнью имеют особый облик и страдают умственной и физической отсталостью.
Синдром Тернера - заболевание, поражающее девочек, характеризуется частичным или полным отсутствием одной или двух Х-хромосом. Заболевание встречается у одной из 3000 девочек. Девочки с таким заболеванием обычно очень маленького роста и у них не функционируют яичники.
Синдром Х-трисомии - заболевание, при котором девочка рождается с тремя Х-хромосомами. Встречается данное заболевание в среднем у одной из 1000 девочек. Характеризуется синдром Х-трисомии незначительной задержкой умственного развития и в некоторых случая бесплодием.
Синдром Клайнфелтера - заболевание, при котором у мальчика имеется одна лишняя хромосома. Заболевание встречается у одного мальчика из 700. Больные синдромом Клайнфелтера, как правило, имеют высокий рост, каких-то заметных внешних аномалий развития нет (после полового созревания затруднен рост волос на лице и несколько увеличены молочные железы). Интеллект у больных обычно нормальный, но часто встречаются нарушения речи. Мужчины, страдающие синдромом Клайнфелтера, обычно бесплодны.
Муковисцидоз - генетическое заболевание, при котором нарушаются функции многих желез. Муковисцидоз поражает людей только европеоидной расы. Приблизительно каждый двадцатый белый человек имеет один поврежденный ген, способный в случае его проявления вызвать муковисцидоз. Заболевание возникает, если человек получает два таких гена (от отца и от матери). В России муковисцидоз, по разным данным, встречается у одного новорожденного из 3500-5400, в США - у одного из 2500. При данном заболевании повреждается ген, отвечающий за выработку белка, который регулирует перемещение натрия и хлора через оболочки клеток. Происходит обезвоживание и увеличение вязкости секрета желез. В результате густой секрет блокирует их деятельность. У больных муковисцидозом плохо усваиваются белок и жир, как следствие - сильно замедляются рост и набор веса. Современные методы лечения (прием ферментов, витаминов и специальная диета) позволяют половине больных муковисцидозом прожить более 28 лет.
Гемофилия - генетическое заболевание, характеризующееся повышенной кровоточивостью вследствие дефицита одного из факторов свертывания крови. Заболевание наследуется по женской линии, при этом поражает в подавляющем большинстве мальчиков (в среднем одного из 8500). Гемофилия возникает, когда оказываются поврежденными гены, отвечающие за активность факторов свертывания крови. При гемофилии наблюдаются частые кровоизлияния в суставы и мышцы, что может в итоге приводить к их значительной деформации (то есть к инвалидности человека). Люди, страдающие гемофилией, должны избегать ситуаций, способных привести к кровотечению. Больным гемофилией нельзя принимать препараты, снижающие свертываемость крови (например, аспирин, гепарин, а также некоторые обезболивающие средства). Для профилактики или прекращения кровотечения больному вводят концентрат плазмы, содержащий большое количество недостающего фактора свертывания.
Болезнь Тея Сакса - генетическое заболевание, характеризующееся накоплением в тканях фитановой кислоты (продукта расщепления жиров). Заболевание встречается в основном среди евреев-ашкенази и канадцев французского происхождения (у одного новорожденного из 3600). Дети с болезнью Тея-Сакса с раннего возраста отстают в развитии, затем у них наступают паралич и слепота. Как правило, больные доживают до 3-4 лет. Методов лечения данного заболевания не существует.
При изучении характера наследования различных признаков у человека описаны все известные типы наследования и все типы доминирования. Многие признаки наследуются моногенно , т.е. определяются одним геном и наследуются в соответствии с законами Менделя. Моногенных признаков описано более тысячи. Среди них есть как аутосомные, так и сцепленные с полом. Некоторые из них приведены ниже.
Моногенные болезни встречаются у 1-2% населения земного шара. Это очень много. Частота спорадических моногенных болезней отражает частоту спонтанного мутационного процесса. Среди них большую долю составляют болезни с биохимическим дефектом. Типичным примером является фенилкетонурия .
Семейное проявление
синдрома Морфана
Это тяжелое наследственное заболевание, обусловленное мутацией одного гена, нарушающей нормальный цикл превращения фенилаланина. У больных эта аминокислота накапливается в клетках. Болезнь сопровождается выраженной неврологической симптоматикой (повышенной возбудимостью), микроцефалией (маленькая голова) и в итоге приводит к идиотии. Диагноз ставится биохимически. В настоящее время в родильных домах проводится стопроцентное скринирование новорожденных на фенилкетонурию. Болезнь излечима, если вовремя перевести ребенка на специальную диету, исключающую фенилаланин.
Еще один пример моногенной болезни — синдром Морфана, или болезнь “паучьих пальцев” . Доминантная мутация одного гена имеет сильный плейотропный эффект. Помимо усиленного роста конечностей (пальцев), у больных наблюдается астения, порок сердца, вывих хрусталика глаза и другие аномалии. Болезнь протекает на фоне повышенного интеллекта, в связи с чем ее называют “болезнью великих людей”. Ею болели, в частности, американский президент А. Линкольн и выдающийся скрипач Н. Паганини.
Многие наследственные болезни связаны с изменением структуры хромосом или их нормального количества, т.е. с хромосомными или геномными мутациями. Так, тяжелое наследственное заболевание у новорожденных, известное как “синдром кошачьего крика ”, вызвано утратой (делецией) длинного плеча 5-й хромосомы. Эта мутация приводит к патологическому развитию гортани, что вызывает характерный плач ребенка. Болезнь несовместима с жизнью.

Широко известная болезнь Дауна является результатом присутствия в кариотипе лишней хромосомы из 21-й пары (трисомия по 21-й хромосоме). Причиной служит нерасхождение половых хромосом при образовании половых клеток у матери. В большинстве случаев появления у новорожденных лишней хромосомы возраст матери достигает, по крайней мере, 35 лет. Мониторинг частоты этого заболевания в районах с сильным загрязнением окружающей среды обнаружил существенное увеличение количества больных этим синдромом. Предполагается также влияние вирусной инфекции на организм матери в период созревания яйцеклетки.
Отдельную категорию наследственных болезней составляют синдромы, связанные с изменением нормального количества половых хромосом . Как и болезнь Дауна, они возникают при нарушении процесса расхождения хромосом в гаметогенезе у матери.
У человека, в отличие от дрозофилы и других животных, Y-хромосома играет большую роль в определении и развитии пола. При отсутствии ее в наборе с любым количеством Х-хромосом особь фенотипически будет женской, а ее присутствие определяет развитие в сторону мужского пола. В частности, особи мужского пола с хромосомным набором ХХY + 44А больны синдромом Клайнфельтера . Они характеризуются умственной отсталостью, непропорциональным ростом конечностей, очень маленькими семенниками, отсутствием сперматозоидов, ненормальным развитием молочных желез и другими патологическими признаками. Увеличение числа Х-хромосом в сочетании с одной Y-хромосомой не изменяет определение мужского пола, а лишь усиливает синдром Клайнфельтера. Впервые кариотип ХХYY был описан в 1962 г. у 15-летнего мальчика со значительной умственной отсталостью, евнухоидными пропорциями тела, с уменьшенными в размере яичками и оволосением по женскому типу. Подобные же признаки характерны для больных с кариотипом ХХХYY.
 Синдром Клайнфельтера (1) и синдром Тернера-Шерешевского (2)
Синдром Клайнфельтера (1) и синдром Тернера-Шерешевского (2)
Отсутствие одной из двух Х-хромосом в кариотипе женщины (ХО) вызывает развитие синдрома Тернера-Шерешевского . Больные женщины обычно низкорослы, менее 140 см, коренасты, со слабо развитыми молочными железами, имеют характерные крыловидные складки на шее. Как правило, они бесплодны из-за недоразвития половой системы. Чаще всего беременность при этом синдроме приводит к самопроизвольному аборту. Только около 2% больных женщин сохраняют беременность до конца.
Трисомия (ХХХ) или полисомия по Х-хромосоме у женщин часто вызывает заболевание, сходное с синдромом Тернера-Шерешевского.
Наследственные болезни, связанные с изменением числа Х-хромосом, диагносцируются цитологическим методом по количеству в клетках телец Барра или полового хроматина. В 1949 г. М. Барр и Ч. Бертрам, изучая интерфазные ядра нейронов у кошки, обнаружили в них интенсивно красящееся тельце. Оно присутствовало только в ядрах клеток самок. Оказалось, что оно встречается у многих животных и всегда связано с полом. Эта структура получила название полового хроматина , или тельца Барра. В ходе тщательного цитологического и цитогенетического анализа было установлено, что половой хроматин представляет собой одну из двух женских половых хромосом, находящуюся в состоянии сильной спирализации и потому неактивную. У женщин с синдромом Тернера-Шерешевского (кариотип ХО) не обнаруживается полового хроматина, так же как и у нормальных мужчин ХY. Нормальные женщины ХХ и аномальные мужчины имеют по одному тельцу Барра, а женщины ХХХ и мужчины ХХХY — по два и т.д.
Лица с наследственными заболеваниями обычно рождаются с большими физическими отклонениями, что позволяет рано диагносцировать болезнь. Но иногда заболевание не дает о себе знать месяцами и даже десятилетиями. Например, тяжелая наследственная болезнь, вызванная поражением центральной нервной системы — хорея Гентингтона — может проявиться только после 40 лет, и тогда ее носитель успевает оставить потомство. Для больных характерны непроизвольные подергивающиеся движения головы и конечностей.
Бывает так, что человек производит впечатление абсолютно здорового индивидуума, но у него есть наследственная предрасположенность к определенному заболеванию, которое проявляется под воздействием внешних или внутренних факторов. Например, некоторым людям свойственна тяжелая реакция на определенные лекарственные препараты, которая обусловлена генетическим дефектом — отсутствием в организме специфического фермента. Иногда наблюдается смертельная реакция на наркоз с виду совершенно здоровых людей, но на самом деле носящих в себе в скрытом виде особую наследственную болезнь мышц. У таких пациентов во время или после операции, проходящей под наркозом, внезапно подскакивает температура (до 42°).