Талассемия – это генетическое заболевание крови. Образуется недостаточное количество гемоглобина, белка, отвечающего за перенос кислорода и углекислого газа. Он состоит из четырех пептидов. 90% молекул гемоглобина содержат два альфа-глобина и два бета-глобина (гемоглобин А или A1), 2.5% содержат вместо бета-цепей дельта-цепи (гемоглобин А2), а остальные представляют собой гемоглобин A, состарившийся в процессе эксплуатации в эритроцитах (гемоглобин A3). При мутации в генах, отвечающих за синтез одного из глобинов, состав гемоглобина нарушается. Эти изменения влекут гибель эритроцитов.
Причины и факторы риска развития заболевания
Талассемия – заболевание наследственное, то есть мутация передается от родителя ребенку. Если кто-то из родственников страдал от этой патологии, риск развития заболевания повышается.
Второй фактор риска – этническая принадлежность. Больше всего талассемия распространена в Африке, Средней Азии и странах Средиземноморья, где она и была открыта (в переводе с греческого “талассемия” - это “морская анемия”). Различают два основных типа талассемии: альфа и бета. В первом случае мутация затрагивает гены, отвечающие за синтез альфа-глобинов, во втором – бета.
Альфа-глобины кодируются четырьмя генами. Тяжесть заболевания будет зависеть от количества патологически измененных участков ДНК:
- 1 измененный ген – бессимптомная форма. Но при ней человек становится носителем заболевания и может передать его своим детям;
- 2 гена – легкое течение болезни;
- 3 гена – тяжелое течение болезни;
- 4 гена – редкий тип заболевания, который плохо совместим с жизнью. Большинство плодов гибнет еще в период внутриутробного развития, а родившиеся малыши, в основном, умирают вскоре после рождения или нуждаются в пожизненной терапии. В отдельных случаях удается их вылечить путем пересадки костного мозга.
Бета-глобины кодируются одним геном, который локализуется в 11-й хромосоме. Если дефектный ген содержится только в одной хромосоме из пары, заболевание протекает легко (малая талассемия). Повреждение обеих хромосом приводит к очень серьезному заболеванию, известному как большая талассемия или болезнь Кули (анемия Кули).
Коды МКБ-10 (Международная классификация болезней 10 пересмотра):
- альфа-талассемия - D56.0
- бета-талассемия - D56.1
- дельта-бета-талассемия - D56.2
- носительство признака талассемии - D56.3
- наследственное персистирование фетального гемоглобина (НПФГ) - D56.4
- другие талассемии - D56.8
- талассемия неуточненная - D56.9
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников - бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Лечение талассемии
Определяется типом и степенью тяжести. При умеренно выраженных симптомах лечение не назначают. Время от времени проводят только переливание крови. В основном это нужно после операций, родов или для предотвращения возможных осложнений. Люди с бета-талассемией требуют более частых переливаний крови. Для нормализации избыточного уровня железа им также назначают специфические препараты, которые связывают и выводят железо.
При выраженной и тяжелой формах болезни существует два способа лечения:
- частые переливания крови (раз в несколько недель), которые сочетают с приемом препаратов, выводящих лишнее железо из организма;
- пересадка костного мозга – единственный метод, который помогает полностью излечить человека от талассемии. К сожалению, далеко не всегда трансплантация бывает успешной.
Лекарственные препараты при талассемии назначают только для коррекции симптомов и осложнений. Медикаментозной терапии самого заболевания не существует.
Осложнения талассемии
Возможные осложнения заболевания:
- излишек железа, которое входит в состав гемоглобина. Накопление этого элемента в организме приводит к поражению сердца, печени, эндокринной системы;
- подверженность инфекциям. Особенно актуально для пациентов, которым удалили селезенку;
- деформация костей, связанная с увеличением объема костного мозга. Чаще всего этот процесс затрагивает кости черепа, реже конечностей. Они истончаются и чаще ломаются;
- спленомегалия - увеличение селезенки, где в основном и гибнут дефектные эритроциты. Если селезенка увеличена очень сильно, ее удаляют. Эта операция называется спленэктомия;
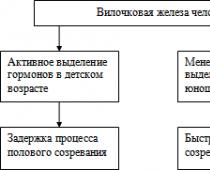
- задержка роста и полового созревания;
- сердечные болезни (хроническая сердечная недостаточность и аритмии) могут развиться при тяжелом течении заболевания.
Правильный образ жизни при заболевании
Существуют простые советы, которые помогают людям с талассемией лучше переносить симптомы заболевания.
- не употреблять никакие витаминно-минеральные комплексы или пищевые добавки, содержащие железо;
- сбалансированное разнообразное питание;
- профилактика инфекционных заболеваний, в первую очередь вакцинация.
Прогноз при талассемии
При легкой форме талассемии прогноз очень хороший. Такие пациенты не требуют постоянного лечения, и осложнения крайне редки. При средней и тяжелой форме прогноз также хороший, но необходимы регулярные переливания крови и прием препаратов, связывающих железо. Такие лекарства очень важны, поскольку наибольшее количество смертей больных талассемией связано с накоплением этого элемента в организме. Пациентам с костными осложнениями может понадобиться операция.
Профилактика талассемии
Талассемия – заболевание наследственное. Поэтому, если один или оба родителя больны, необходимо проконсультироваться с врачом-генетиком на этапе планирования беременности.
Талассемии представляют собой гетерогенную группу гемоглобинопатии, в основе которых лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина А. Талассемия - это мишеневидноклеточная анемия с нарушенным соотношением НЬА и HbF по биохимическим показателям; при этом возможна частичная недостаточность определенной цепи или ее полное отсутствие при преобладании другой цепи. Так, при нарушении синтеза ß-цепи будут преобладать а-цепи и наоборот. Бета-талассемия обусловлена снижением продукции ß-цепей гемоглобина. Неповрежденные а-цепи избыточно накапливаются в клетках эритропоэза, что ведет к повреждению мембраны и разрушению как клеток эритроидного ряда в костном мозге, так и эритроцитов в периферической крови; развиваются неэффективный эритропоэз и гемолиз с гипохромией эритроцитов, ибо содержание гемоглобина в эритроцитах недостаточно. Первыми описали ß-талассемию американские педиатры Кули и Ли в 1925 г. Тяжелая гомозиготная форма ß-талассемии получила название болезни Кули, или большой талассемии. Кроме того, по выраженности анемии и других клинических симптомов выделяют промежуточную, малую и минимальную талассемию. Помимо стран Средиземноморья талассемия встречается во Франции, Югославии, Швейцарии, Англии, Польше, а также у жителей Закавказья и Средней Азии, где в некоторых регионах частота носительства достигает 10-27 %.
Что провоцирует / Причины Талассемии:
При талассемии нарушается синтез одной из четырёх цепей глобина. Наследование патологии от одного (гетерозиготность) или обоих родителей (гомозиготность), тип нарушенной цепи определяют выраженность клинических проявлений. Причины повышенной гибели эритроцитов связаны с нарушенной структурой клетки из-за неправильного соотношения цепей глобина в гемоглобине. Кроме укорочения жизни эритроцитов при данном заболевании происходит гибель клеток предшественников эритроцитов в костном мозге.
Патогенез (что происходит?) во время Талассемии:
Патогенез ß-талассемии связан с мутацией в локусе ß-глобина на 11-й паре хромосом, нарушающей синтез ß-глобиновой цепи. Вследствие неадекватного синтеза гемоглобина развивается гипохромная анемия. Преципитаты избыточного количества а-цепей удаляются из эритроцитов и эритрокариоцитов клетками ретикулогистиоцитарной системы; при этом клетки повреждаются и быстрее разрушаются. Таков механизм неэффективного эритропоэза и гемолиза эритроцитов и ретикулоцитов; гибель последних происходит в селезенке. При ß-талассемии накапливается также HbF, обладающий большим сродством к кислороду; однако отдача его тканям затруднена, что приводит к их гипоксии. Неэффективный эритропоэз способствует расширению плацдарма кроветворения, что отражается на структуре скелета; вместе с тем деструкция эритрокариоцитов в костном мозге ведет к повышенному всасыванию железа и патологической перегрузке организма железом. Гематологические признаки ß-талассемии иногда выявляются у больных анемией среди русских.
Симптомы Талассемии:
Клиника большой талассемии проявляется уже в детстве. У больных детей своеобразный башенный череп, монголоидное лицо с увеличенной верхней челюстью. Ранний признак болезни Кули - сплено- и гепатомегалия, развивающиеся за счет экстрамедуллярного кроветворения и гемосидероза. Со временем у них формируются цирроз печени, сахарный диабет в результате фиброза поджелудочной железы, а гемосидероз миокарда приводит к застойной сердечной недостаточности. Гомозиготная бета-талассемия (большая талассемия, анемия Кули) характеризуется резким снижением образования HbA1, значительным увеличением содержания HbF, низким, нормальным или повышенным содержанием НbA2. Содержание НbF может колебаться от 30 до 90 %, иногда бывает ниже 10%. Течение заболевания характеризуется тяжелой гемолитической анемией, проявляющейся к концу первого года жизни ребенка, гепато- и спленомегалией, монголоидиостью лица и башенным черепом, отставанием ребенка в физическом развитии, нередко желтушностью и бледностью кожных покровов. У части больных развиваются язвы в области голеней. Рентгенологически обнаруживают симптом «ежика» или «щетки», который положителен при увеличении содержания HbF, отрицателен при увеличении процента НbA2. У детей в возрасте от 6 мес. до 1 года в мелких костях стоп и кистей выявляется истончение коркового слоя со вздутием кости и образованием грубосетчатой структуры костного мозга. Начиная с 1-го года жизни ребенка отмечается нарушение развития костей, быстро прогрессирующее до периода полового созревания. Длительно продолжающийся гемолиз (ретикулоцитоз, увеличение свободной фракции билирубина сыворотки крови, уробилинурия, гиперсидеремия), частые переливания эритроцитной массы приводят к развитию гемосидероза печени и селезенки. Нередко происходит образование билирубиновых камней в желчных путях. Уровень гемоглобина достигает 30-50 г/л, цветовой показатель 0>5 и ниже. В мазках крови обнаруживают мишеневидные эритроциты, отличающиеся малым содержанием гемоглобина и укорочением продолжительности жизни, анизопойкилоцитоз, эритро- и нормобласты. Отмечается повышение осмотической стойкости эритроцитов, лейкопения (в период гемолитического криза). В костном мозге - раздражение эритро-нормобластического ростка. Иногда возникают апластический криз или явления гиперспленизма. При тяжелой гомозиготной талассемии больные умирают на первом году жизни, при сравнительно более спокойной форме заболевания они могут дожить до взрослого возраста. Гетерозиготная бета-талассемия протекает в виде как бессимптомной, так и манифестной форм с незначительно увеличенной селезенкой, специфическими костными изменениями, нередко выраженной гипохромной анемией, часто анизоцитозом, пойкилоцитозом и мишеневидностью эритроцитов, повышенной их осмотической резистентностью увеличением количества НbA2 (примерно до 8% от общего гемоглобина), у части больных - HbF (до 5%). При гетерозиготной дельтабета-талассемии (F) отмечается высокое содержание HbF при нормальном уровне НbA2. Клинические признаки и гематологические сдвиги аналогичны встречающимся при гетерозиготной бета-талассемии. Гомозиготные формы дельтабета-талассемии (F) проявляются почти теми же клинико-гематологическими нарушениями, что и гомозиготная бета-талассемия. У больных с этой формой заболевания обнаруживается только HbF. Среди больных талассемией удается выделить лиц с гетеро-и гомозиготными формами А2F-талассемии, которые по признакам, характеризующим их течение, по существу мало отличаются от бета-талассемии. В группе больных бета-талассемией случаи большой талассемии с выраженными клиническими проявлениями встречаются реже, чем промежуточные и малые формы. При обследовании родственников больных чаще обнаруживается минимальная форма бета-талассемии. Выделяют следующие формы а-талассемии: водянка плода с гемоглобином Bart"s (у4). гемоглобинопатия Н (бета4), а-талассемия-1 и а-талассемия-2. Водянка плода представляет собой гомозиготное состояние (по генам а-th-l), несовместимое с жизнью. Беременность в подобных случаях непроизвольно прерывается и у плода выявляют водянку мозга, гепатомегалию. Электрофоретическим исследованием гемоглобина обнаруживается Hb Bart"s (80-90%, сочетающийся со следами НbН. Гемоглобинопатия Н - один из вариантов а-талассемии - проявляется гемолитической анемией, увеличением селезенки, тяжелым течением костных изменений. Картина периферической крови характеризуется понижением содержания гемоглобина, анизо- и пойкилоцитозом, гипохромией и множественными включениями в эритроцитах (выпавший в осадок гемоглобин Н). Гетерозиготные формы а-талассемии выявляются у родственников больных гемоглобинопатией Н. а-Талассемия-1 (малая форма заболевания) возникает при сочетании гена а-th-l с нормальным геном a-цепочкового синтеза. Она характеризуется небольшой анемией, умеренным анизо- и пойкилоцитозом, внутриэритроцитарными включениями, повышенной осмотической резистентностью эритроцитов. У взрослых больных а-талассемией-1 гемоглобиновые фракции бывают в пределах нормы, у новорожденных выявляется Hb Bart"s (5-10%). а-Талассемия-2 (минимальная форма заболевания) развивается при сочетании гена a-th-2 с нормальным геном а-цепочкового синтеза. Клинические проявления отсутствуют.
Диагностика Талассемии:
При анализе крови определяется гипохромная гиперрегенераторная анемия разной степени тяжести. В мазке крови обнаруживают гипохромные эритроциты малых размеров, мишеневидные, различной формы; много нормоцитов. В биохимическом анализе крови выявляются гипербилирубинемия за счет свободной фракции, гиперсидеремия, снижение ОЖСС, повышение активности ЛДГ. В эритроцитах повышен уровень фетального гемоглобина. Альфа-талассемия распространена преимущественно в Юго-Восточной Азии, Китае, Африке и в Средиземноморье. Синтез а-цепей кодируют 4 гена, поэтому степень нарушения их синтеза меньше, чем при ß-талассемии; выраженный дисбаланс развивается только тогда, когда поражены все 4 гена. В то же время агрегаты из ß-цепей, количество которых при а-талассемии обнаруживают в избытке, более растворимы, чем агрегаты из а-цепей, поэтому гемолиз при а-талассемии выражен слабее, чем при ß-талассемии, а эритропоэз более эффективен. Следовательно, клинические и лабораторные данные при а-талассемии выражены менее отчетливо, чем при ß-талассемии; их основное отличие в биохимическом составе гемоглобина эритроцитов: при а-талассемии уменьшено содержание а-цепей гемоглобина. Пренатальная (дородовая) диагностика Если оба родителя страдают талассемией, целесообразно исследование плода в период беременности на предмет заболевания талассемией с целью возможного своевременного прерывания беременности. Обнаружение т.н. «гомозиготных» (более тяжелых) форм талассемии у плода– показание для прерывания беременности. Применяется 2 основных метода- фетоскопия и амниоцентез. Оба они связаны с получением клеток плода с помощью пункции через переднюю брюшную стенку (первая из них делается под контролем УЗИ) с последующим медико-генетическим исследованием полученных клеток. Врач-генетик определит предпочтительный метод исследования в зависимости от срока беременности, данных УЗИ и индивидуальных особенностей беременной. Оба метода имеют свой риск, в первую очередь - преждевременные роды. Имеется также, хотя и очень небольшой, риск присоединения инфекции и даже гибели плода (по данным литературы- около 3%). Для решения вопроса о планировании семьи людям, имеющим родственников, больных талассемией, обязательно следует обращаться к генетику и он назначит Вам, если нужно, необходимое дородовое обследование.
Лечение Талассемии:
Переливания эритроцитов. При тяжелых формах талассемии потребность в переливаниях эритроцитарных препаратов крови возникает уже с первых месяцев жизни и сохраняется, хотя и в разной степени, пожизненно – развивается так называемая трансфузионная зависимость. Это означает, что гемоглобин в крови больных постоянно продолжает снижаться и других реальных способов его повышения, кроме таких переливаний, нет. Желательно, чтобы в крови больного содержание гемоглобина не падало до низких цифр, лучше осуществлять повторное переливание при еще удовлетворительных его уровнях- 95-100г/л. Дело в том, что при выраженном снижении гемоглобина активизируются многие присущие именно талассемии патологические процессы: например, упоминавшееся избыточное патологическое костеобразование, увеличение размеров печени и селезенки; ухудшается функция всех органов, снижается сопротивляемость к инфекциям из-за усилившегося кислородного голодания. При большой b-талассемии кроме замещения недостатка эритроцитов в циркулирующем кровяном русле с помощью переливаний эритроцитарных компонентов крови достигаются подавление собственного избыточного, но малоэффективного кровообразования в костном мозге больного, а также уменьшается всасывание железа в кишечнике. Таким образом, в наблюдении за больным талассемией важным является недопущение развития эпизодов выраженного падения уровня гемоглобина – это, во-первых, непосредственно может угрожать жизни, а во-вторых, способствует прогрессированию патологических проявлений талассемии. В то же время переливание эритроцитарных препаратов крови имеет свои существенные минусы. Как известно, при переливаниях препаратов крови обязательно учитывается совместимость донора и реципиента по группе крови, резус фактору. Но поскольку не существует в природе генетически одинаковых людей, то при повторных переливаниях организм пациента раньше или позже начинает вырабатывать белки-антитела, реагирующие с другими, более сложными частями мембран переливаемых кровяных телец (эритроцитов). Поэтому через некоторое время (обычно 3-4 года) организм больного становится биологически «совместимым» уже не с любым донором, подходящим по группе крови и резус-фактору, а только с определенными донорами, обладающими специфическим набором белков-антигенов на эритроцитах. Поэтому переливания эритроцитарных сред при талассемии желательно проводить по индивидуальному подбору, осуществляемому специальной изосерологической лабораторией станций переливания крови. Кроме того, эритроцитарные среды для больных талассемией должны быть специально очищенными от других биологических компонентов, содержащихся в крови (лейкоциты, многочисленные белки плазмы), поскольку они являются причиной т.н. «пирогенных» реакций, проявляющихся ознобами и повышениями температуры, нередко до высоких цифр. Цельную кровь в настоящее время не переливают, переливания необработанной дополнительными методами эритроцитной массы также не желательны. В качестве высокоочищенных эритроцитарных сред сейчас используются размороженные, отмытые или фильтрованные эритроциты, которые гораздо реже вызывают реакции. Но как бы то ни было, при наличии трансфузионной зависимости переливания неизбежны, поэтому можно лишь принимать меры к уменьшению их побочных действий и предупреждению реакций и осложнений. При более легких формах талассемии, когда у больных либо имеется анемия легкой степени (на уровне 90-110г/л), либо гемоглобин в норме, а главное – что он стабильно сохраняется у больного с течением времени и не продолжает неуклонно снижаться, переливания препаратов крови не проводятся. Десферал. Важной частью лечения является выведение избытка железа из организма с помощью препаратов из группы «хелатов» (т.н. «хелатная терапия»), осуществляемая препаратом «десферал». В настоящее время принято лечение подкожными многочасовыми инъекциями, наиболее удобно применение специальных аппаратов - т.н. помп, которые прикрепляются к одежде. Из фиксированного в помпе шприца десферал за несколько часов постепенно вводится подкожно пациенту. В идеале больные тяжелой формой талассемии должны получать десферал на протяжении всей жизни по 5 дней в неделю, но в реальной жизни это пока труднодостижимо. В странах с большой распространенностью талассемии, особенно высокоразвитых (например, в Италии) существуют специальные государственные программы помощи больным талассемией, которые предусматривают, кроме другого лечения, обеспечение необходимым для них десфералом и помпами для его введений. Из стран бывшего СССР аналогичная программа существует в Азербайджане. Хранить десферал следует в темном месте при +8-15˚С, разводить непосредственно перед началом введения. С осторожностью назначается лечение десфералом у детей младше 2–х лет ввиду относительно большего риска развития побочных эффектов лечения. У таких детей лечение десфералом начинают, если уже проведено около 15-20 переливаний препаратов крови, т.е. потребность в переливаниях уже довольно большая. Для улучшения качества жизни больных предпочтительна инфузия десферала ночью. Следует систематически менять места подкожных инъекций во избежания местного повреждения кожи и подлежащих мягких тканей. Как и любое лечение, терапия десфералом имеет свои возможные побочные эффекты. Чаще встречаются аллергические реакции на препарат, возможна также лихорадочная реакция. В случае появления на фоне лечения десфералом каких-либо новых жалоб следует обратиться к врачу, он решит вопрос о продолжении лечения и о том, каким образом лечить побочные эффекты десферала. Удаление селезенки (спленэктомия) У некоторых больных очень большие размеры селезенки сами по себе начинают негативно сказываться на состоянии гемоглобина и других показателей системы крови. В таких случаях проводят ее хирургическое удаление. От самой талассемии эта операция не излечивает, хотя и может смягчить ее проявления (что, впрочем, может и не произойти). Спленэктомия проводится только при очень больших размерах селезенки, а также когда имеют место явные признаки ее патологического действия на другие показатели крови (так называемый «гиперспленизм»), Операция не целесообразна ранее достижения возраста 5 лет, оптимальным считается возраст 8-10 лет. Первый год обычно наблюдается хороший эффект, но затем возможно рецидивирование проявлений талассемии, может нарасти увеличение печени. Кроме того, возрастает инфекционный риск, особенно относительно присоединения т.н. «пневмококковой» инфекции в виде сепсиса, пневмонии. В связи с этим обязательна вакцинация против пневмококка, проводимая предпочтительно в предоперационный период. Вообще же решение вопроса об удалении селезенки всегда должно приниматься с большой осторожностью. Пересадка (трансплантация) костного мозга В настоящее время все большее распространение получает лечение талассемии с помощью пересадки костного мозга. Это единственный метод радикального лечения талассемии. При выявлении талассемии желательно, чтобы пациенты и члены их семей были «типированы по системе HLA» (т.е. прошли довольно сложное биологическое обследование на совместимость) с целью поиска возможного донора костного мозга. Однако найти подходящего донора обычно сложно, сама процедура поиска совместимого неродственного донора пока остается дорогой и длительной по времени. Очень дорога и сама пересадка костного мозга. Близкие родственники, даже если являются совместимыми по антигенам HLA, сами нередко имеют талассемию. Поэтому реальными кандидатами на лечение пересадкой костного мозга пока еще становятся относительно немногие больные талассемией. Следует отметить, что результаты пересадки костного мозга во многом зависят от качества проводившейся больному ранее терапии. Лучше результаты пересадки костного мозга у детей. Хотя разработка и внедрение новых методов лечения талассемий, включая пересадку костного мозга, продолжается, все же для подавляющего большинства больных реально возможными пока остаются приводившиеся выше «традиционные» методы лечения. В настоящее время также разрабатываются методы лечения талассемии с помощью генной инженерии. Больным талассемией следует соблюдать диету (стол №5). Полезны напитки, содержащие танин: чай, какао, а также орехи, соя – эти продукты уменьшают всасывание железа. Из-за склонности к кариесу рекомендуются фтористые зубные пасты, своевременная санация полости рта. Для улучшения функции печени врач назначает лекарства – т.н. «гепатопротекторы». К ним относится липоевая кислота, вит Е, препараты типа хорошо известного «эссенциале». Улучшает выведение железа из организма аскорбиновая кислота (витамин С) в дозе 50мг/сутки до 10 лет и 100мг/сутки у детей старше 10 лет. Также применяется лечение витаминами группы В, фолиевой кислотой. Увеличение дозы витаминов проводят при стрессе, беременности. Назначаются курсы желчегонных трав - мята, овес, кукурузные рыльца, барбарис, а также тюбажи. Особенности лечения других форм талассемии «Промежуточная» талассемия. Вследствие более мягкого течения заболевание не требует постоянных переливаний - обычно не чаще 1 раза в 2-3нед – 2-3мес. При присоединении интеркуррентных заболеваний, при операциях- проводятся переливания эритроцитарных сред при уровне гемоглобина ниже 70 г/л. Как правило, назначается лечение десфералом, но предпочтительнее после специального исследования обмена железа по уровню его содержания в крови и ответу на разовое введение десферала (т.н. «десфераловый тест»). При больших размерах селезенки с признаками избыточно повышенной функции рассматривается вопрос об ее оперативном удалении «Малая» талассемия. Переливаний эритроцитов не требует. При анемии назначают фолиевую кислоту, также возможно решение вопроса о лечении десфералом по уровню сывороточного железа или десфераловому тесту. При тяжелых формах талассемии, особенно с наличием трансфузионной зависимости, больному оформляется инвалидность.
Профилактика Талассемии:
Профилактика талассемии основывается на выявлении подверженных риску лиц посредством программ скрининга носителей или изучения историй семьи и предоставления адекватной информации о риске и о возможностях сокращения такого риска. Бета-талассемия обладает уникальным свойством: здоровых носителей можно определять простым, недорогим и точным анализом крови. Таким образом, можно выявлять пары носителей и информировать о генетическом риске до того, как они создадут семью. Скрининг - это недорогой и доступный способ выявления носителей, который можно предлагать в широкой гамме ситуаций в различных условиях: в средней школе, перед вступлением в брак или в женских консультациях. Пары выявленных таким образом носителей информируются о генетическом риске и о возможных вариантах его снижения, которые обычно включают дородовую диагностику. Большинство пар, подверженных риску талассемии, обращаются за дородовой диагностикой гемоглобинопатии. Стандартный метод диагностики - это взятие пробы ворсинок хорионов и анализ ДНК при сроке беременности 10-12 недель. Программы скрининга и консультирование могут привести к серьезному сокращению подверженных болезни новорожденных. Эти программы можно анализировать благодаря беседам с родителями, имеющими больных детей, хотя у них был доступ к скринингу и консультированию. В большинстве случаев рождение затронутых болезнью детей является результатом неспособности систем здравоохранения адекватно информировать родителей о возможном риске и мерах профилактики, а не тем, что они отвергают тестирование плода. Выбор соответствующей стратегии для введения профилактики талассемии зависит от конкретных условий. В некоторых обществах начинать можно с обеспечения дородовой диагностики для тех пар, которые уже знают о существовании риска либо благодаря скринингу, либо потому, что у них больной ребенок. Такой подход значительно сокращает число новорожденных с заболеванием. С другой стороны в обществах, где дородовая диагностика еще не обеспечена, можно предложить скрининг для лиц репродуктивного возраста. Эта стратегия ведет к меньшему сокращению числа детей с заболеванием, однако обычно стимулирует спрос на службы дородовой диагностики. В настоящее время во многих странах имеются примеры эффективного применения методов профилактики талассемии на основе различных программ скрининга носителей. Например, в Греции, на Кипре, в Исламской Республике Иран и Италии скрининг на талассемию до вступления в брак является стандартной; большинство пар, подверженных риску, определяется достаточно своевременно, чтобы предложить раннюю диагностику на первой беременности. Большинство таких пар пользуются этой услугой и имеют здоровых детей. В Соединенном Королевстве Великобритании и Северной Ирландии и других странах северо-западной Европы, где дородовая диагностика широко распространена, скрининг предлагается во время беременности. Программы скрининга нуждаются в поддержке в виде просвещения общественности и регламентарных структур, с тем чтобы отдельные лица могли принимать обоснованные решения и чтобы люди были защищены от дискриминации на основании результатов их тестов. Некоторые национальные программы, цель которых - содействие скринингу носителей, в свою очередь стимулировали социальные изменения, включая принятие во многих странах прекращения беременности в тех случаях, когда доказано, что плод страдает серьезным генетическим расстройством. Это привело к разработке соответствующих технологий и служб в Бахрейне, Исламской Республике Иран и Саудовской Аравии. Масштабы принятия и внедрения программ профилактики расширяются во многих частях Азии, например в Индии, Индонезии, Китае, Малайзии, на Мальдивских Островах, в Сингапуре и Таиланде. Генетическое консультирование играет важную роль в защите автономии индивидуума или пары и в осуществлении ими своего права на максимальную информацию о болезни и имеющихся возможностях. Эффективность служб по борьбе против талассемии зависит от учета их сотрудниками культурной практики и принятия действий, соответствующих данному социальному контексту. При консультировании также необходимо учитывать культурные, религиозные и этические взгляды личности или пары. Успех генетического консультирования в значительной степени определяется его просветительным, добровольным и непредписывающим характером.
К каким докторам следует обращаться если у Вас Талассемия:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Талассемии, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Euro lab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом . Клиника Euro lab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны . Посмотрите детальнее о всех услугах клиники на ее .
(+38 044) 206-20-00
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни . Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача , чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации , возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой . Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе . Также зарегистрируйтесь на медицинском портале Euro lab , чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм:
| B12-дефицитная анемия |
| Анемии, обусловленные нарушением синтеза утилизацией порфиринов |
| Анемии, обусловленные нарушением структуры цепей глобина |
| Анемии, характеризующиеся носительством патологически нестабильных гемоглобинов |
| Анемия Фанкони |
| Анемия, связанная со свинцовым отравлением |
| Апластическая анемия |
| Аутоиммунная гемолитическая анемия |
| Аутоиммунная гемолитическая анемия |
| Аутоиммунная гемолитическая анемия с неполными тепловыми агглютининами |
| Аутоиммунная гемолитическая анемия с полными Холодовыми агглютининами |
| Аутоиммунная гемолитическая анемия с тепловыми гемолизинами |
| Болезни тяжелых цепей |
| болезнь Верльгофа |
| Болезнь Виллебранда |
| болезнь Ди Гулъелъмо |
| болезнь Кристмаса |
| Болезнь Маркиафавы-Микели |
| Болезнь Рандю - Ослера |
| Болезнь тяжелых альфа-цепей |
| Болезнь тяжелых гамма-цепей |
| Болезнь Шенлейн - Геноха |
| Внекостномозговые поражения |
| Волосатоклеточный лейкоз |
| Гемобластозы |
| Гемолитико-уремический синдром |
| Гемолитико-уремический синдром |
| Гемолитическая анемия, связанная с дефицитом витамина Е |
| Гемолитическая анемия, связанная с дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ) |
| Гемолитическая болезнь плода и новорожденного |
| Гемолитические анемии, связанные с механическим повреждением эритроцитов |
| Геморрагическая болезнь новорожденных |
| Гистиоцитоз злокачественный |
| Гистологическая классификация лимфогранулематоза |
| ДВС-синдром |
| Дефицит К-витаминзависимых факторов |
| Дефицит фактора I |
| Дефицит фактора II |
| Дефицит фактора V |
| Дефицит фактора VII |
| Дефицит фактора XI |
| Дефицит фактора XII |
| Дефицит фактора XIII |
| Железодефицитная анемия |
| Закономерности опухолевой прогрессии |
| Иммунные гемолитические анемии |
| Клоповое происхождение гемобластозов |
| Лейкопении и агранулоцитозы |
| Лимфосаркомы |
| Лимфоцитома кожи (болезнь Цезари) |
| Лимфоцитома лимфатического узла |
| Лимфоцитома селезенки |
| Лучевая болезнь |
| Маршевая гемоглобинурия |
| Мастоцитоз (тучноклеточный лейкоз) |
| Мегакариобластный лейкоз |
| Механизм угнетения нормального кроветворения при гемобластозах |
| Механическая желтуха |
| Миелоидная саркома (хлорома, гранулоцитарная саркома) |
Талассемия возникает при мутации генов, которые отвечают за синтез пептидов - основных элементов гемоглобина. Тяжесть болезни зависит от количества мутировавших генов. Талассемия приводит к дефициту гемоглобина - белка, играющего ключевую роль в транспортировке кислорода и углекислого газа. Молекулы гемоглобина содержат по два альфа-глобина и бета-глобина. Генная мутация приводит к изменению состава крови и последующей гибели эритроцитов.
Талассемия вызывает следующие последствия в организме:
- возникает излишек железа в гемоглобине, из-за чего нарушается работа сердца, печени и эндокринной системы;
- нарушается иммунитет, и организм становится более подверженным инфекциям;
- развиваются сердечные болезни (аритмия и хроническая сердечная недостаточность);
- увеличивается объем костного мозга, что приводит к деформации костей черепа и конечностей;
- увеличивается в размерах селезенка, где в основном и гибнут эритроциты (в тяжелых случаях увеличенную селезенку удаляют операционным путем);
- наблюдается задержка роста и полового созревания.
Талассемия является наследственным заболеванием, то есть передается ребенку от родителей. Риск развития болезни повышается, если ранее она наблюдалась у кого-либо из ближайших родственников. Также фактором риска становится этническая принадлежность. В наибольшей степени талассемия распространена в странах Средиземноморья, Средней Азии и Африки.
Выделяют две основных разновидности талассемии: альфа и бета. Первая из них подразумевает мутацию генов, отвечающих за синтез альфа-глобинов, а вторая - бета-глобинов. Тяжесть заболевания и его основные особенности будут зависеть от типа пораженных генов, а также от количества патологических участков ДНК. Учитывая, что альфа-глобины кодируются четырьмя различными генами, выделяют следующие разновидности болезни:
- Один измененный ген - бессимптомная форма. Человек является носителем заболевания и может передать его по наследству.
- Два гена - легкое течение заболевания.
- Три гена - тяжелое течение заболевания.
- Четыре гена - редкий тип талассемии, чаще всего приводящий к летальному исходу. Большинство плодов с генетическими изменениями гемоглобина погибают еще до рождения, а появившиеся на свет младенцы умирают в течение нескольких последующих дней либо обретают болезнь пожизненно. В редких случаях ситуацию спасает пересадка костного мозга от здорового человека с идентичной структурой ДНК и составом крови.
Что касается бета-глобинов, они кодируются одним геном, локализующемся в 11-й хромосоме. При наличии дефектного гена лишь в одной из парных хромосом развивается легкая степень заболевания - малая талассемия. Если поражены обе хромосомы, возникает серьезное заболевание - большая талассемия (болезнь Кули).
Симптомы талассемии
Современные медицинские технологии позволяют выявлять талассемию еще в процессе дородовой диагностики. В этом случае будущей матери может назначаться особое лечение, способное устранить патологию до рождения ребенка. У родившихся детей признаки талассемии проявляются в зависимости от типа заболевания и числа мутаций. В одних случаях симптомы регистрируются сразу после рождения, а в других - в течение первые двух лет жизни.
Выделяют следующие распространенные симптомы заболевания:
- бледность либо желтушность кожи;
- замедление роста;
- потемнение мочи;
- деформация костей, в особенности костей черепа;
- увеличение живота.
Диагностика талассемии
Чаще всего симптомы, характерные для талассемии, не позволяют уверенно поставить диагноз. Для этого необходимо пройти специальное обследование. В первую очередь пациент сдает общий анализ крови, по которому становится заметным снижение количества эритроцитов, появление более мелких, разных по размеру и форме клеток. Помимо зрелых клеток в крови могут присутствовать их предшественники - бласты.
Также назначаются прочие специфические анализы крови, позволяющие определить степень тяжести заболевания. Могут назначаться и особые молекулярные тесты (ПЦР), которые помогают выявить мутации на генном и клеточном уровне. Помимо анализа крови требуется диагностика печени и селезенки - органов, в наибольшей степени страдающих от талассемии. Кроме того, необходимо проверить состояние костной ткани. Для этого проводятся УЗИ и рентгенография.
Крайне важно постараться диагностировать у ребенка талассемию еще на этапе его вынашивания, особенно если один или оба родители, ранее были больны этим заболеванием либо являются его потенциальными носителями. В этом случае применяются такие методы диагностики, как биопсия ворсинок хориона (на 11-ой неделе беременности) и отбор околоплодных вод - амниоцентез (назначают на 16-й неделе).
Лечение заболевания
При постановке такого диагноза, как талассемия, терапия назначается в зависимости от стадии и степени тяжести заболевания. Если симптомы выражены слабо либо отсутствуют вовсе, лечение не проводится. Пациент нуждается только в периодическом переливании крови, особенно после операционных вмешательств и родов, а также для предотвращения различных осложнений. При этом людям с бета-талассемией требуются более частые переливания крови, а для нормализации повышенного уровня железа в организме они вынуждены принимать специфические препараты, связывающие и выводящие это вещество.
Если болезнь протекает в ярко выраженной или тяжелой форме, применяются следующие способы лечения:
- Частые переливания крови (каждые несколько недель), сочетаемые с приемом медикаментов, выводящих из организма лишнее железо.
- Пересадка костного мозга - наиболее эффективный метод лечения талассемии, позволяющий полностью избавиться от заболевания при благоприятном исходе. Трансплантация - серьезная операция, которая не всегда бывает успешной.
- Прием специальных лекарственных препаратов, корректирующих симптомы и осложнения.
Прогноз при талассемии и профилактика осложнений
Легкая форма талассемии имеет достаточно хороший прогноз. Таким пациентам не требуется постоянное лечение, и у них крайне редко возникают осложнения. Средняя и тяжелая формы заболевания тоже имеют благоприятный прогноз, однако для этого пациент должен проходить регулярные переливания крови и употреблять связывающие железо препараты.
В большинстве случаев смертельный исход наступает при избыточном накоплении железа в организме, а также нарушении работы жизненно важных органов. Однако даже с негативными последствиями можно справиться посредством проведения операции. Как показывает практика, большинство людей, следующих указаниям врача, живут вполне обычной жизнью.
Если был поставлен такой диагноз, как талассемия, очень важно ответственно подходить к вопросу о создании семьи. В случае беременности будущая мать должна проходить анализ крови и УЗИ по особому расписанию, чтобы как можно раньше убедиться в наличии либо отсутствии болезни у плода. В остальном рекомендуется избегать приема любых витаминно-минеральных комплексов или пищевых добавок, которые содержат железо. Также пациентам назначается сбалансированное и разнообразное питание. Наконец, они обязаны вовремя проводить профилактику инфекционных заболеваний посредством вакцинации.
Основанием для патологии служит изменение структуры генов, отвечающих за цепные соединения глобина. Подвергнувшийся мутации ген может передаваться от родителей ребёнку. В итоге у заболевшего бета-цепи или не образуются вовсе, или синтезируются в недостаточном объёме.
Вместе с тем продуцирование остальных цепочек, содержащихся в глобине, продолжает осуществляться. В итоге формируются нестабильные комплексы белковых веществ, которые разрушающее действуют на кровяные клетки - эритроциты. Затем у пациента диагностируется анемия, и начинается процесс отложения железа в органах и тканях.
Симптомы
Симптоматика довольно разнообразна. Зависит в основном от формы патологии.
- Изменение в строении черепа, он приобретает практически квадратную форму.
- Переносица приобретает седловидную форму.
- Глаза сужаются.
- Верхняя челюсть увеличивается и становится больше нижней.
- Печень, а также селезёнка увеличиваются в размерах.
- Кожный покров бледнеет, приобретает желтушный оттенок.
- Язвенные болячки в области голеней.
- Наличие билирубиновых камней в желчных путях.
- Развитие водянки плода, обычно, данная патология заканчивается смертью эмбриона в утробе.
- Явное отставание и на физическом, и на половом уровне.
- Вялость, быстрая утомляемость.
- Сниженный иммунитет.
Диагностика талассемии у ребёнка
Специалист осуществляет общий осмотр, уточняет данные о наследственности. Далее назначает анализ крови. Гемоглобин снижается до 30-50 единиц.
Помимо перечисленных мероприятий, осуществляют ещё ряд манипуляций.
- Специалист берёт мазок крови.
- Назначают биохимическое исследование крови.
- В отдельных случаях показа пункция костного мозга.
- Нередко используют ПЦР диагностику на уровне молекулярной биологии.
- Электрофорез вещества на ацетат-целлюлозной пленке.
- Рентген костей.
- Обычно требуется консультация детского гематолога, медгенетика.
Осложнения
Чем опасна данная проблема крови? Взрослым, столкнувшимся с такой патологией у ребёнка, нужно понимать, что болезнь может привести к летальному исходу.
Также не исключены ряд заболеваний в будущем: цирроз печени, на фоне дисфункции поджелудочной железы развитие сахарного диабета, сердечной недостаточности.
Лечение
Что можете сделать вы
Родителям следует выполнять все назначения специалиста. Это касается, прежде всего, необходимой сдачи всех анализов, только тогда эксперт сможет определить точную схему лечения и приступить к немедленной терапии.
Также необходимо проконтролировать соблюдение специальной диеты. Таким детям нужно употреблять чай, орехи, сою и какао. Перечисленная продукция минимизирует всасывание железа. Кроме того, ребенку нужно давать витамин С. Элемент помогает выведению железа из органов и тканей. Для оптимизации деятельности печени следует принимать гепатопротекторы, курс около месяца.
Что делает врач
После постановки диагноза специалист принимает ряд мер для оказания помощи ребёнку. Вид терапии зависит от стадии патологии и данных, полученных благодаря исследованиям.
- Сейчас наиболее действенным специалисты считают процесс переливания специально подготовленных эритроцитов, которые гораздо реже оказывают негативные эффекты, вместе с длительным вводом медпрепаратов.
- При острейших протеканиях болезни (в частности, при развитии β-талассемии) гемотрансфузия цельной крови (кровь переливается целиком, то есть жидкость вместе с белками и кровяными клетками) или эритроцитарной части (только эритроцитов) обеспечивают только кратковременный эффект, кроме того, при них повышается риск возникновения гемосидероза (отложение вещества, который содержит оксид железа).
- Медецинские препараты необходимо вводить на протяжении нескольких часов подкожно. Для этой цели применяют специализированные устройства, их можно прикрепить к одежде. Пациент с тяжёлым характером заболевания в получении средства нуждается пять дней в неделю. Курс не ограничен. То есть на протяжении всей своей жизни ребёнок должен получать необходимый ему препарат. Инъекции делать следует в различные места, чтобы не возникло повреждение тканей. При обнаружении гемолитических кризов показано введение глюкокортикоидов в минимальных дозировках.
- При увеличенных размерах селезенки прибегают к спленоэктомии. Хирургическое вмешательство противопоказано пациентам до пяти лет. Рекомендованный возраст - с 8 до 10 лет. Положительный эффект отмечают в первый год после операции, во многих случаях после года снова наблюдается ухудшение. Также повышается риск инфекционных заболеваний.
- Также современные эксперты в этой области осуществляют трансплантацию костного мозга. Но проблема здесь - в поиске подходящего донора, а также в вопросах очереди и стоимости операции.
Профилактика
Предотвращение патологии начинают ещё в дородовый период. У беременных, которые находятся в группе риска, специалисты исследуют эмбрион на наличие данной патологии. Если родители будущего малыша страдают любой формой заболевания, специалисты не исключают вынужденного прерывания беременности. Специалисты используют две методики: осмотр самого плода и исследование околоплодных вод. Используя данные способы, эксперты получают клетки плода, путём проведения пункции через переднюю брюшную стенку. Описываемые способы являются крайне негативными, поскольку могут привести к ранним родам или выкидышу, инфицированию и даже смерти эмбриона.
Чтобы не допустить патологии малыша, взрослым, у которых есть близкие с такой проблемой, надлежит проконсультироваться до планирования пополнения в семье со специалистом-генетиком, чтобы тот начал соответствующее дородовое обследование.
Гематолог
Высшее образование:
Гематолог
Самарский государственный медицинский университет (СамГМУ, КМИ)
Уровень образования - Специалист
1993-1999
Дополнительное образование:
«Гематология»
Российская Медицинская Академия Последипломного Образования
Бета-талассемия — это собирательный термин, под которым скрывается ряд генетически обусловленных заболеваний крови. Данное патологическое состояние встречается достаточно редко. Для того чтобы проявились его характерные симптоматические проявления, оба родителя должны передать ребенку неполноценный ген. Это патологическое состояние может проявляться как в легкой форме, не ведущей к явному ухудшению состояния здоровья малыша, так и в крайне тяжелой. В настоящее время это заболевание уже достаточно хорошо изучено. При подтверждении диагноза родителям ребенка необходимо морально подготовиться к длительной борьбе с проявлениями этого патологического состояния.
Стоит отметить, что сейчас это генетическое заболевание не может быть полностью излечено, но правильная поддерживающая терапия позволяет значительно улучшить качество и продолжительность жизни больных. Прогноз при этом заболевании зависит от тяжести течения болезни. Смертность детей, страдающих от бета-талассемии, в младенческом и подростковом возрасте крайне высока. Таким образом, при тяжелых формах болезни требуется продолжительное медицинское лечение, предполагающее многие манипуляции, направленные на устранение имеющихся нарушений.

Что такое бета-таласемия?
Впервые это патологическое состояние было описано в 1925 году, когда американские врачи-педиатры проводили обследование детей эмигрантов, являющихся выходцами из Италии. В ряде случаев была выявлена примерно одинаковая картина, включающая признаки малокровия, увеличение селезенки и печени. В дальнейшем появились научные работы, в которых было дано описание легкого течения этого патологического состояния.
Термин «талассемия» был введен только в 1936 году. Дословно название этого заболевания переводится, как «болезнь мокрого побережья». Кроме того, именно в этом году было выдвинуто предположение, что это патологическое состояние является результатом нарушения продукции гемоглобиновых цепочек. После исследований все медицинское сообщество узнало, что такое талассемия и к каким последствиям ведет это патологическое состояние.
У представителей славянских народностей данное заболевание встречается крайне редко. Наиболее часто оно диагностируется у выходцев из стран Срединоземноморья, а кроме того, некоторых африканских национальностей и латиноамериканцев. В настоящее время существует теория, что подобная мутация появилась у людей, проживающих на определенной местности, как адаптационный защитный механизм против малярийного плазмодия. Однако когда имеющаяся у двух родителей рецессивная мутация передается ребенку, это приводит к тяжелым последствиям.
Этиология и патогенез развития бета-талассемии
В настоящее время известно, что у здоровых людей гемоглобин состоит из особых альфа и бета цепей. Чтобы этот элемент нормально функционировал, его выработку и дальнейшую работу должны контролировать локусы в генетическом коде, в том числе 11 и 14 хромосомы. В процессе мутации этих элементах закладывается неправильная информация. Она в дальнейшем становится причиной продукции кровяных телец с явным гемоглобиновым дефектом. Это ведет к фатальным последствиям.
В настоящее время известно, что гемоглобин является единственным веществом в организме человека, который может связывать молекулы кислорода и переносить их от легких к отдаленным тканям. Нарушение структуры вырабатываемого гемоглобина из-за имеющейся генетической мутации становится причиной стремительного развития анемии, в результате которой все ткани не получают жизненно важный для них кислород. Бета-талассемия проходит несколько этапов развития. Сначала наблюдается нарушение синтеза одной фракции бета-цепи. Это запускает компенсаторные явления. Таким образом, фракции альфа-цепи продуцируются в избыточном количестве. Из-за их стремительного увеличения наблюдается развитие окислительных процессов. Разрушение избыточных альфа-цепей приводит к разрушению мембран эритроцитов. Изменения строения красных кровяных телец становятся причиной нарушения их функции.

В настоящее время известно, что из-за дефекта в строении гемоглобина эритроциты становятся крайне чувствительными. Они быстро разрушаются при соприкосновении со стенками сосудов или при столкновении друг с другом. Из них полностью выходит гемоглобин. В результате этого процесса разрушаются не только сформировавшиеся эритроциты, но и их предшественники, то есть еще не созревшие элементы крови. Далее образуются высвобождающиеся пигменты, которые начинают сначала насыщать плазму, а затем откладываться в тканях различных органов. Учитывая, что из разрушенных эритроцитов высвобождается большое количество железа, этот микроэлемент провоцирует нарушение работы в клетках различных органов и становится причиной серьезных нарушений.
При тяжелом течении последствия подобных изменений становятся фатальным. Места пораженных клеток во всех жизненно важных органах быстро замещаются соединительной тканью, не способной выполнять необходимые функции замещенных участков. Помимо всего прочего, эритроциты, имеющие дефект, не могут полноценно выполнять свою функцию, то есть насыщать клетки организма кислородом. Это только усугубляет ситуацию, поэтому патологические изменения стремительно нарастают. Постепенно снижается функция почти всех жизненно важных органов.
Классификация бета-талассемии
Этот термин является достаточно расплывчатым, и под ним скрывается сразу несколько генетических групп заболеваний, которые сопровождаются тяжелыми формами анемии. Именно из-за этого существует множество подходов к классификации бета-талассемии. В зависимости от выраженности клинических проявлений это патологическое состояние может иметь следующие формы:
- бессимптомную;
- легкую;
- среднюю;
- тяжелую.
В некоторых случаях это заболевание протекает без каких-либо признаков. В других — в течение нескольких дней после рождения ребенка нарастающие нарушения на фоне тяжелейшей анемии становятся причиной гибели больного. Помимо всего прочего, в зависимости от выраженности клинических проявлений может быть выделена малая, большая и промежуточная бета-талассемия. Каждая форма имеет свои характерные признаки и механизмы их развития.
Одна из классификаций этого патологических состояний учитывает путь передачи дефектных генов ребенку. Согласно этому принципу выделяется гомозиготная и гетерозиготная разновидности бета-талассемии. Определить это не всегда удается. Гомозиготная форма подтверждается только в случае, если мутировавший ген имеется у обоих родителей. Эта разновидность заболевания является наиболее распространенной. Гетерозиготная бета-талассемия наследуется только от одного родителя. В настоящее время процессы передачи мутивровших генов при развитии этого заболевания уже хорошо изучены.

В большинстве случаев оба родителя являются носителями этого патологического гена, но при этом не болеет сами. При формировании генокода ребенка ему достается мутировавший ген от обоих родителей. В то же время, если один из родителей болеет легкой или даже тяжелой формой бета-талассемии, а второй не является носителем мутировавшего гена, риск развития этого патологического состояния у ребенка минимален. В этом случае здоровый ген от одного из родителей полностью вытесняет мутировавший. Таким образом, гетерозиготная бета-талассемия в настоящее время встречается крайне редко.
Существуют перинатальные тесты, позволяющие определить наличие этого патологического состояния, а также его форму еще в период внутриутробного развития. Гомозиготная талассемия является показанием для прерывания беременности. Обычно плод погибает в последнем триместре или же через несколько недель после рождения.
Клинические проявления бета-талассемии
Лишь в редких случаях бессимптомного течения имеющиеся признаки не позволяют в раннем возрасте определить у ребенка наличие этого заболевания. Как правило, когда речь идет о бета-талассемии, симптомы во многом зависят от формы течения болезни. Таким образом, в некоторых случаях проявления будут крайне слабыми, а в других очень тяжелыми. Достаточно часто диагностируется большая бета-талассемия. Эта форма патологии получила название лихорадки Кули, протекает крайне тяжело и сопровождается появлением серьезных нарушений. В большинстве случаев симптомы очевидны уже в первые дни жизни малыша. Своего апогея они достигают примерно к 6 месяцам. К характерным признакам развития бета-талассемии относятся:
- снижение показателей гемоглобина до 60-20г/л;
- тяжелая анемия;
- пожелтение кожи;
- сильная бледность и даже синюшность;
- нарушение формирования костной ткани;
- изменения формы лица и черепа;
- замедление развития;
- увеличение печени и селезенки;
- частые инфекционные и воспалительные заболевания;
- проявления вторичного геморрагического синдрома.

У большинства детей, которые не погибают в первые полгода жизни, на фоне развития большой бета-талассемии наблюдается появление тяжелых нарушений работы эндокринной системы. Может проявиться карликовость и задержка полового созревания. Прогноз при этой форме заболевания крайне неблагоприятный и даже при частых переливаниях крови и выполнении других медицинских процедур продолжительность жизни больных редко превышает 20 лет. Примерно 30% детей умирают в первый год жизни. Большая часть больных доживает при правильном лечении до 8 лет.
Кроме того, достаточно распространенной является малая бета-талассемия. В этом случае выявляется мутация исключительно в 11-й паре хромосом. В результате этого у больных наблюдается лишь легкая анемия, причем уровень гемоглобина лишь незначительно понижен. К характерным клиническим проявлениям малой формы бета-талассемии относятся:
- расширение эритроцидного ростка в костном мозге;
- микроцитоз;
- низкое содержание гемоглобина в эритроцитах;
- анизопойкилоцидоз;
- незначительное увеличение селезенки.
Нередко эта форма болезни протекает без каких-либо серьезных физиологических изменений. При малой талассемии у детей нет проявлений, отражающихся на общем состоянии здоровья. Возможна легкая анемия, так как количество эритроцитов незначительно ниже нормы. Кроме того, в результатах анализов крови может обнаруживаться повышенный уровень железа и некоторые другие вещества, указывающие на наличие проблемы. В этом случае дети нормально развиваются, но при этом диагностика крайне важна, так как в дальнейшем уже взрослым людям необходимо планировать семью и рождение детей, чтобы их потомки были здоровыми.
Промежуточная бета-талассемия подтверждается, когда у больного в одном гене имеется слабая мутация, а в другом — тяжелая. В этом случае признаки болезни достаточно размыты, и нельзя на 100% причислить ее ни к большой, ни к малой форме бета-талассемии. Как правило, у детей с первых дней жизни наблюдается пожелтение кожных покровов, но уровень гемоглобина находится на нижней границе нормы. При таком течении патологии гемотрансфузию нужно делать достаточно редко, так как характерных признаков анемии не наблюдается.
В некоторых случаях может выявляться сильно повышенный уровень железа, что является показанием для удаления селезенки. Больному требуется постоянное наблюдение со стороны врача, так как вполне возможен переход бета-талассемии к более тяжелому течению. При таком варианте патологии больным очень важно придерживаться здорового образа жизни, так как вредные привычки могут спровоцировать ухудшение состояния.
Осложнения при бета-талассемии
Тяжелые формы заболевания, к примеру, анемия Кули, предрасполагает к появлению ряда тяжелых нарушений. К распространенным осложнениям относится гемосидероз. К характерным проявлениям этого патологического состояния, развивающего на фоне бета-талассемии, относится:
- специфический зеленоватый цвет кожных покровов;
- прогрессирующий сахарный диабет;
- некроз поджелудочной железы;
- фиброз и цирроз печени;
- сбои в работе сердца.
У большинства больных в дальнейшем наблюдается развитие синовита крупных суставов, что сильно отражается на возможности нормально передвигаться. В большинстве случаев талассемия — болезнь, протекающая крайне тяжело. Нарушение роста костей предрасполагает к появлению переломов. Кроме того, к распространенным осложнениям бета-талассемии относятся цирроз печени, фиброз поджелудочной железы, кардиосклероз, трофические язвы на ногах, воспаления в легких, нарушения полового развития, нарастающая сердечная недостаточность, холецистит, пневмонии и сепсис. Кроме того, может иметь место нарушение умственного и физического развития.

Методы диагностики бета-талассемии
В большинстве случаев характерные симптомы патологического состояния достаточно отчетливо проявляются уже в первые дни жизни малыша, поэтому сразу же педиатр может назначить консультации у ряда узконаправленных специалистов и проведение необходимых исследований. Заболевание талассемия нередко диагностируется при проведении генетических перинатальных тестов, поэтому родители, которые получили ранее исчерпывающую информацию о состоянии ребенка до его рождения, уже готовы к борьбе за его здоровье. В некоторых случаях это патологическое состояние выявляется по имеющимся у младенца признакам уже после рождения. Когда речь идет о таком заболевании, как талассемия, диагностика обычно предполагает проведение таких исследований, как:
- определение уровня гемоглобина;
- пункция костного мозга;
- УЗИ брюшной полости;
- рентгенография;
- краниография.
Оценка состояния ребенка, а также получение данных от различных анализов и исследований позволяет быстро определить характер имеющегося заболевания. Помимо всего прочего, определение формы течения патологического состояния позволяет понять, какие именно последствия она будет иметь.
Как проводится лечение бета-талассемии?
Подход к терапии того патологического состояния во многом зависит от тяжести его течения. Всем больным требуется специальная диета, направленная на снижение всасывания кишечником железа. В случае бессимптомного или легкого течения повышение потребления чая, орехов, сои, кофе, какао позволяет значительно сократить поступление в организм этого микроэлемента.
При промежуточной и большой бела-талассемии требуются регулярные переливания крови. Частота таких процедур зависит от выраженности симптоматических проявлений. Переливание крови позволяет добиться временного результата. В некоторых случаях подобные процедуры сопряжены с появлением некоторых побочных эффектов. Кроме того, при таком патологическом состоянии, как талассемия, лечение требует применения препаратов, способствующих выведению железа из организма. Обычно с этой целью используется Десферал. Это лекарство является прекрасным профилактическим средством, позволяющим не допустить такое опасное осложнение, как сидероз.
При стремительном ухудшении состояния из-за гемолитического криза может быть показана терапия большими дозами глюкокортикоидами. При значительном увеличении селезенки и постоянно повышенном уровне содержания железа рекомендовано выполнение скленэктомии. Это оперативное вмешательство лучше выполнять, когда ребенок достигает 8-10 лет. Обычно после удаления селезенки наступает значительное улучшение состояния. Однако даже такая кардинальная мера позволяет лишь на определенное время устранить имеющиеся симптоматические проявления.
Самым действенным способом лечения бете-талассемии является пересадка костного мозга, что осложняется высокой стоимостью подобных операций в сочетании со сложностью подбора подходящего донора. В развитых странах Европы существуют государственные программы и благотворительные фонды, которые позволяют больным бета-талассемией получать необходимое лечение. Наилучшими донорами становятся близкие родственники. Несмотря на значительный положительный эффект, который может быть достигнут при трансплантации костного мозга, имеются определенные возрастные ограничения для ее проведения. Большинство врачей рекомендуют такую операцию детям старше 5 лет. Это значительно повышает шансы на благоприятный исход.
Народные средства от бета талассемии
При этом генетическом заболевании требуется направленное медицинское лечение. Народные средства в этом случае могут выступать в качестве дополнительных. Они при определенных формах этого заболевания могут поспособствовать снижению выраженности симптоматики. Для увеличения количества кровяных телец детям может быть рекомендован свежий свекольный сок с добавлением небольшого количества меда. Для улучшения состояния больного достаточно принимать по 1 стакану средства 3 раза в день.
Помимо всего прочего, определенную пользу может принести сок подорожника. Его необходимо принимать по 1 ч.л. перед едой. Еще один народный рецепт против проявлений бета-талассемии предполагает употребление смеси свежих соков моркови, редьки и свеклы. Такое средство нужно принимать 3 раза в день по 2 ст.л.
Профилактика бета-талассемии
В настоящее время существует немало способов перинатальной диагностики генетических заболеваний. В случае если родители входят в группу риска, может быть рекомендовано проведение биопсии хориона или амниотической жидкости. Это позволяет выявить патологию у плода на ранних стадиях его развития. В случае если женщина или мужчина знают о том, что они являются носителями дефектного гена или страдают бета-талассемией в легкой или бессимптомной форме, им необходимо очень тщательно подходить к выбору партнера.

Беременность должна планироваться. Перед зачатием паре нужно провести генетические тесты, чтобы рассчитать возможность появления больного ребенка. В случае если в процессе беременности было подтверждено, что плод имеет бета-талассемию, которая в дальнейшем будет протекать в тяжелой форме, большинство врачей рекомендуют аборт.
Родители могут сами решать, как им лучше поступить в этом случаев. Рекомендации врачей вполне оправданы, так как в дальнейшем, скорее всего, произойдет выкидыш, который на позднем сроке может быть травматичным для женщины, или же на свет появится ребенок с нарастающими нарушениями работы органов. Талассемия, что проявляется тяжелой симптоматикой, в большинстве случаев ведет к летальному исходу.